Aim: Next Generation Sequencing (NGS) offers superior HLA genotyping resolution compared to other typing methods. However, certain regions of the HLA genes, e.g. homopolymers, microsatellites, and GC rich regions, remain at high risk of base call error. We have developed an approach for assessing sequence quality whereby we plot the percentage of the 2nd most frequent base that contributes to a consensus base call at every sequence position analyzed for all loci. Identification of poor-quality regions enable focused analysis strategies that minimize the risk of genotyping errors.
Method: We sequenced 11 HLA genes on Illumina’s MiSeq platform using an NGS target enrichment assay. Reads underwent analysis with TypeStream Visual Analysis Software to calculate the percentage of a second base call (PSB) at each base position of every locus. In the absence of nonspecific regions, homozygous alleles should have a PSB of 0%, with no alternative base calls, while heterozygous ones should have a PSB of 50%. Regions which are challenging to sequence or align can cause PSB to deviate from 0% or 50% and may result in unreliable base calling. To identify regions at high risk of consensus base call error, we calculated the PSB at each position for every locus.
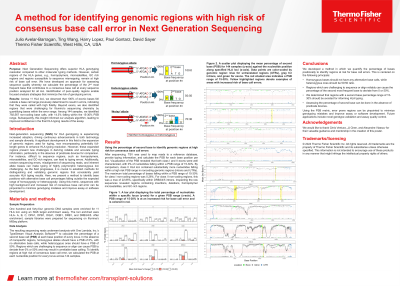
Results: Across the 11 HLA loci, we observed that 100% of exonic bases fell outside a PSB range of 15-30%, a range previously determined to result in base call errors. This indicated that all bases were called with high fidelity. Beyond exons, we also identified regions that were challenging for Illumina’s sequencing chemistry by mapping bases with a PBS of 15-30%. Among 144 samples, we identified 792,567 non-coding base calls, with 14.3% falling within the 15-30% PSB range. These regions primarily consisted of homopolymer and microsatellites within introns. Subsequently, this insight informed our analysis algorithm, leading to improved confidence in the final HLA typing results of the assay.
Conclusion: We detail a method to identify base positions with alternative base call percentages falling outside the expected range of homozygosity or heterozygosity. Using the PSB metric, sequences with high background and increased risk of consensus base call error can be pinpointed to minimize genotyping mistakes and improve assay or software development. Future applications include novel genotype validation and assay quality control.